Aims
- To evaluate the entire NGS and bioinformatics workflow
Outcomes
- Standardisation of VCF files
- Identification of missed variants, leading to a change in capture kit used
- Implementation of a gender check for all subsequent testing
- A new reference set for benchmarking variant callers
The Customer
UZ Leuven is Belgium’s largest university hospital with almost 10,000 employees. In 2023, the hospital performed over 776,000 consultations, had around 185,000 day admissions, and carried out nearly 58,000 surgical procedures. The staff carried out over 125,000 functional tests for cardiology, respiratory diseases, endoscopy and neurophysiology, and over 16 million laboratory tests.
That’s over 44,000 laboratory tests a day…
In terms of genetic testing, the Centre for Human Genetics UZ Leuven, KU Leuven (CHG Leuven) performs a wide variety of tests, some of them based upon next generation sequencing (NGS) of samples, such as exome sequencing or whole genome sequencing. On a yearly basis, they perform over 100,000 genetic tests. The exact test used depends on the clinical question or patient phenotype. CHG Leuven is also the reference centre for exome or genome testing of particular diseases for clinics local to Leuven and around Belgium.
The Challenge
With such high sample throughput comes great responsibility.
A false negative or false positive genetic test could have serious implications for the patient and their treating clinician.
So, while rigorous validation of tests and novel technologies are often performed in-house, unforeseen technical issues could creep in at any stage of the testing pipeline.
Therefore, technical EQA schemes are invaluable for evaluating workflows by detecting potential errors or possible improvements in sample preparation, the test employed, the sequencing platform and the bioinformatic computational analysis pipelines used for data analysis.
CHG Leuven evaluates their entire NGS and bioinformatics workflow with technical EQA each year, but in 2021 they also wanted to compare two different variant callers.
The EQA Scheme Used
In 2021 CHG Leuven enrolled in the germline Single Nucleotide Variants (SNVs) and small insertions/deletions (indels) regular EQA as well as the Copy Number Variants (CNVs) scheme from EMQN.
Still a pilot scheme when they enrolled, the SNVs and indels scheme is now an ISO accredited scheme (ISO 17043:2012) applicable to any genomic testing laboratory using NGS as part of their germline testing pipeline. It aims to assess the analytical and technical process of genotyping performed in a customer’s particular diagnostic testing environment while assessing raw data quality.
This EQA is suitable for any NGS-based approach including single gene, panel testing, whole exome sequencing, or whole genome sequencing and assesses variants of 50 bp or less in size.
The Process
EMQN provided CHG Leuven with one DNA sample of approximately 2 µg DNA. After sequencing and processing, they could submit up to three result files which allowed them to assess different tests along with various aspects of their wet laboratory and bioinformatics pipeline.
The diagnostic laboratory processed the EQA samples from start to finish with their routine NGS workflow. They used the results of the EQA samples to compare two different variant callers with their in-house bioinformatics pipeline.
After Leuven sent the analysis files to EMQN, we performed our internal assessment and sent a comprehensive individual data quality report and variant consensus analysis report for each sample submission to CHG Leuven. We also sent performance evaluations and summary reports that include additional information from the cohort of participants including geographical spread, methodologies employed, common errors, learning points and scheme statistics, allowing participants to benchmark their results.
Our reports also indicated detailed information about the chromosomal variant position, whether the variant is a SNV or an indel, the gene, the laboratory genotype, and the EQA consensus genotype.
The EQA report contains different outcomes for variant detection depending on the EQA scheme used.
- Agree – This means your test has detected the same variant as the one specified by the EQA. No action is needed.
- Missing – This suggests that your test failed to detect a variant even though the EQA confirms it is present (false negative). This is a cause for concern and requires further investigation.
- Disagree – This indicates that the variant called by your laboratory and computational analysis differs from that specified by the EQA. This also suggests there is an issue with your analysis pipeline and requires careful assessment to see where the error originated.
- Extra – This refers to variants detected by your laboratory that other laboratories participating in the EQA scheme did not detect. You can use this information to see if you can reduce the number of false positives by using different variant callers or by modifying your pipeline.
- Not assessed – This refers to variants for which the EQA did not have enough information to make a consensus call. Such variants are disregarded from performance evaluation.
Variant position
1:11949898
7:56019598
1:43921556
X:2738220
14:99175206
Type
snp
indel
snp
snp
indel
Gene
PLOD1
PSPH
ST3GAL3
CD99
BCL11B
Submitted genotype
C/T
G/G
GCTC/G
EQA genotype
C/T
AC/A
G/G
A/G
EQA consensus ratio
46/46
13/35
25/30
33/36
Classification
Agree
Not Assessed
Missing
Disagree
Extra
Table 1. Example of the variant classification output from the EQA scheme.
The Outcomes
1. Standardising file formats
One unforeseen benefit of participating in the CNV EQA scheme for CHG Leuven was to identify issues with bioinformatic file formats. This led to standardisation of file formats for more streamlined use in subsequent schemes, while saving time and resources. It empowers the use of standardised data formats and raises standards.
After the completion of sample preparation and sequencing to identify genetic variants, UZ Leuven’s bioinformaticians performed the routine bioinformatics analysis of the EQA test samples and attempted to submit the resulting ‘Variant Call Files’ (VCF) to the EQA scheme.
VCF files are a standard text file format used in bioinformatics for storing sequence variations discovered by the variant callers. Downstream analyses rely on these VCF files, and they are an essential component required for submission to the EQA process, alongside mandatory BED files and optional FASTQ and BAM files. Together, these files are used to create a consensus call set used to assess the performance of a laboratory.
Dr. Souche discovered that the particular VCF files they routinely used for downstream analysis were not compatible with EQA.
“There were some things in our VCF files that weren’t as they should have been and we had to correct this so that we could successfully participate in the EQA. So EQA helps to standardise file formats as well.” – Dr. Erika Souche
After correcting these VCF files, the laboratory could participate seamlessly in future EQA schemes.
2. Identification of missed variant
The ability of healthcare professionals to reliably detect genetic variants is one of the foundations of diagnostic NGS laboratories. These laboratories use many different variant detection kits to assess CNVs, SNVs, and other genetic variants, depending on the clinical question or patient symptoms presented, but these kits cover hundreds to thousands of variants requiring extensive independent validation not always practical for diagnostic laboratories.
Dr. Souche and colleagues included the EQA sample provided by EMQN in their routine NGS variant detection pipeline. However, the results of the EQA indicated a ‘Missing’ outcome for one particular variant (see box 1 for the definition of ‘Missing’).
The investigation of this false negative result revealed that the capture kit they used did not cover an exon containing the variant of interest, so the variant was missed with their analysis (Figure 1).
“This is a variant that will never be called because there is no coverage there. This information is very important because the diagnostic team must know that the exon is not covered.” – Dr. Erika Souche
Variant position
1:43921556
Type
snp
Gene
ST3GAL3
Submitted genotype
EQA genotype
G/G
EQA consensus ratio
25/30
Classification
Missing -> FN
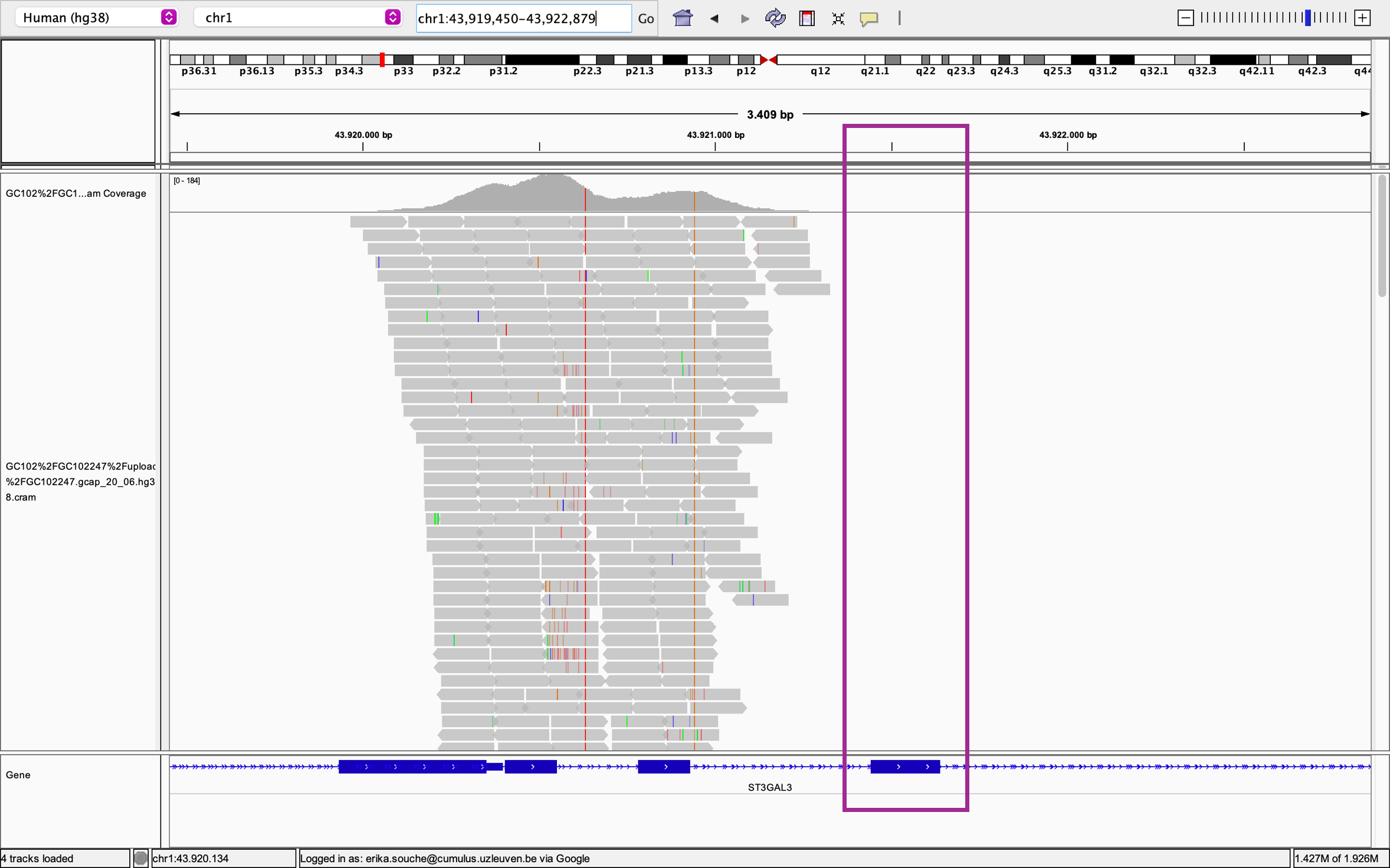
Figure 1. EQA output and Integrated Genome Viewer screenshot showing the coverage of several exons of the ST3GAL3 gene on chromosome one. No sequencing reads were mapped to the exon of interest as shown by the light blue box and led to a ‘Disagree’ call in the EQA results. Horizontal dark blue line and boxes indicate introns and exons of ST3GAL3 respectively.
By identifying this missed variant, thanks to participation in the EQA, the results drove action and led the diagnostic laboratory to change the variant capture kit to one that could robustly detect the variant of interest to improve clinical interpretations.
2. Identification of missed variant
Patient gender plays an important role in variant detection, owing to the presence of one Y chromosome and one X chromosome in genetic males and of two X chromosomes in genetic females (excluding chromosome anomalies). Each of these chromosomes can host independent genetic variants and their detection can be affected by laboratory and bioinformatic pipelines.
When participating in any EQA scheme, CHG Leuven would anonymise the EQA sample to make sure that it is treated the same as any other diagnostic sample. The EQA samples are thus entered into their database. During that process, they are randomly assigned a gender. One of the EQA samples provided by EMQN was randomly assigned as male without any gender check taking place.
This random gender assignment affected how the downstream bioinformatic variant-calling pipelines treated the EQA sample. With their original pipeline, no heterozygous calls are allowed on the X chromosome for male samples, since they only have one copy of chromosome X. This meant that any X chromosome variants detected would be classed as homozygous.
CHG Leuven submitted an X chromosome variant as homozygous for the EQA but for this they received a ‘Disagree’ classification (See box 1). This occurred because 33 of 36 other participants detected a heterozygous variant as the EQA sample was actually female and should have been treated differently in the bioinformatics pipeline.
“If we had performed a gender check on that sample, it would have triggered a warning that there was something wrong with this sample and then we would have been able to correct this.” – Dr. Erika Souche
As a result, UZ Leuven has now implemented a gender check for all subsequent testing to avoid any future issues with misassignment and incorrect sex chromosome variant detection.
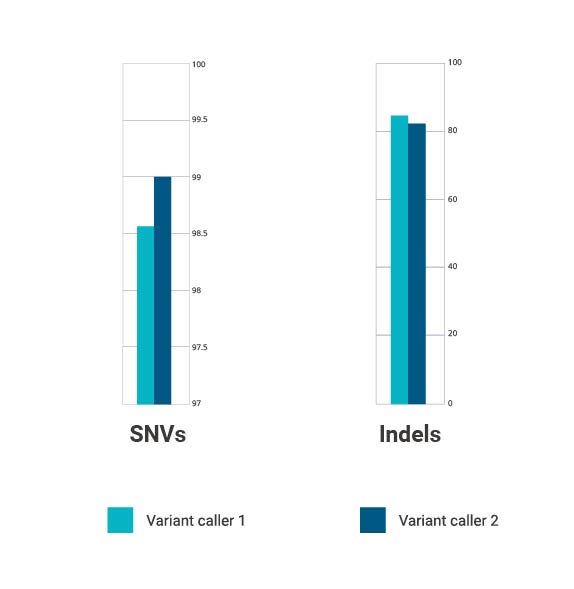
Figure 2. Bar charts comparing the performance of two variant callers where the F score is the mean of precision and sensitivity
Overall, the better performance of variant caller two for the detection of SNVs provided confidence for CHG Leuven to implement this alternative method for all future NGS testing and is now driving more robust, cost-effective, and efficient variant calling for higher standards in the clinic.
Raising standards thanks to EQA
Overall, CHG Leuven passed this EQA scheme and gained accreditation as a sufficient number of variants were successfully called. However, Dr. Souche and colleagues still utilised the scheme as an opportunity to learn crucial information about how they could raise standards and improve their entire pipeline from laboratory to data analysis.
“If all of the laboratories participating in EQA perform this exercise to improve their pipeline, the standards will rise because the next version of EQA will be even better. It will ensure that every laboratory is performing as well as possible.” – Dr. Erika Souche
By taking a deep dive into the results and data provided via EMQN as part of the technological assessment EQA, the team identified areas of improvement including the standardisation of data files, variants undetected with the previous capture kit, implementation of a gender check, and optimisation of the variant caller used.
Ultimately, the EQA raised the standards of NGS variant testing to improve clinical decision making with inevitable benefits for patients in need, all while saving time and money thanks to the streamlining of laboratory and bioinformatic pipelines.